
図表1
バイオシミラーは、先行バイオ医薬品と同等/同質の品質・安全性・有効性を有し、しかも経済性にも優れた医薬品である。2025年6月時点で、国内で上市しているバイオシミラーは19成分である。しかし成分によってはまだ普及の進んでいないものもある。
このため2026年診療報酬改定でも、バイオシミラーを普及させるための診療報酬上のインセンテイブが設けられた。本項ではあたらためてバイオシミラーとは何か、そしてそのさらなる使用促進策について考えていこう。
1 バイオ医薬品とは?
まず本題に入る前に、バイオ医薬品とは何かについて振り返ってみよう。2000年ごろより医薬品の世界は大きく変貌する。それまでは医薬品といえば、化学合成によって作られる低分子化合物が主役だった。それが2000年ごろよりバイオ医薬品が主役へと躍り出る。バイオ医薬品はこれまで治療が困難だったがん、糖尿病、慢性関節リウマチなどの難病に著しい効果をしめした。日本でも肺がんに対するオプチーボの絶大な効果が人々を驚かせた。
バイオ医薬品とは遺伝子組換えや細胞培養等のバイオテクノロジーを応用して作られる医薬品である。バイオ医薬品は、遺伝子組み換え操作によって作られた大腸菌、酵母、チャイニーズハムスターの卵細胞などのいわゆる「細胞工場」によって製造される。このためバイオ医薬品はこれまでの化学合成される低分子の医薬品に比べて、たんぱく製剤であるため分子量も巨大で、構造も複雑だ。しかもその開発や製造には多額の経費を要する。このためその薬価も高額だ。
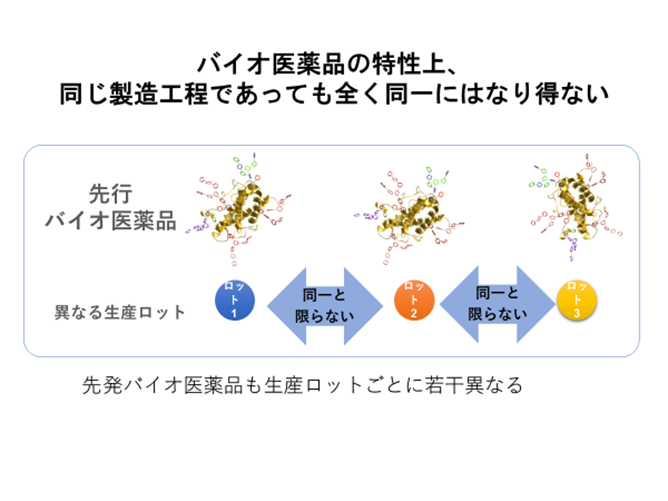
さてこうして新薬メーカ―で作られるバイオ医薬品は先行バイオ医薬品とも呼ばれる。この先行バイオ医薬品についてまず見ていこう。先述したようにバイオ医薬品は生きている細胞工場によって作られることもあり、先行バイオ医薬品の製造工程でも完全に同一のモノを作ることはできない。実際にはロットごとに少しずつ差異がでてくるのだ(図表1)
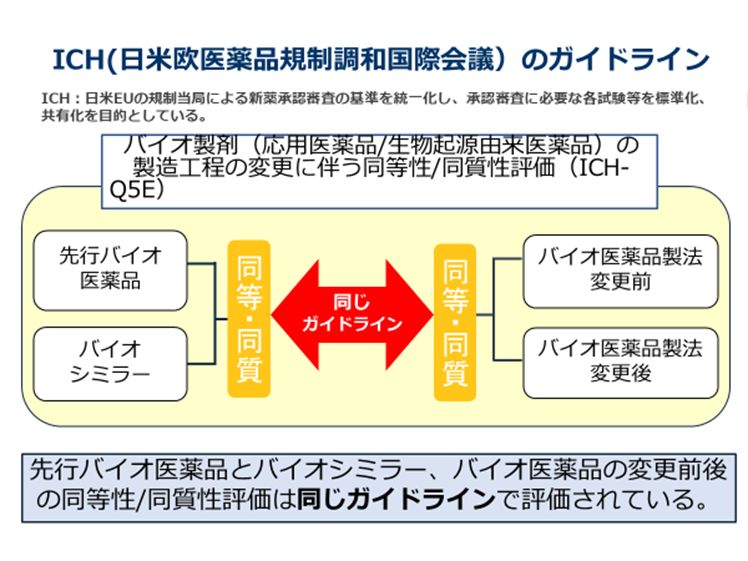
また先行バイオ医薬品も他の医薬品同様、市販後その生産効率の向上や、製剤の品質向上を目的として、製造工程の変更を行うことも多い。こうした製造工程の変更時に製剤の特性や生物活性に変化が生じることがある。たとえば製造工程の変更前後で、塩基性製剤の割合や糖鎖構造、生物活性などが変化することも知られている。このため先行バイオ医薬品の製造工程変更前後の品質の同等性・同質性は、日米欧医薬品規制調和国際会議におけるICH Q5Eというガイドライン1)にしたがって決められている。
このガイドラインによれば、先行バイオ医薬品の製造工程の変更にともなう同等性・同質性の定義とは以下の通りである。「同等性・同質性とは、必ずしも変更前および変更後の製品の品質特性が全く同じであることを意味するものではなく、変更前後の製品の類似性が高いこと、ならびに、品質特性に何らかの差異があったとしても、既存の知識から最終製品の安全性や有効性には影響を及ぼさないであろうことが十分に保証できることを意味する」。
このように同じ製薬企業の同じ製造ラインにおいて製造される先行バイオ医薬品においても、ロット間差異や製造工程変更後の差異が生じる。このことを認めた上で、その最終製品の安全性や有効性を、ある基準の範囲の中で保証することになっている。
2 バイオシミラーとは?
では、次にバイオシミラー(バイオ後続品)について見ていこう。バイオシミラーの定義とは「バイオ後続品の品質・安全性・有効性確保のための指針」2)によれば、以下のように定義される。「国内で既に新有効成分含有医薬品として承認されたバイオテクノロジー応用医薬品(先行バイオ医薬品)と同等・同質の品質、安全性及び有効性を有する医薬品として、異なる製造販売業者により開発される医薬品である」。この場合の同等性とは、先行バイオ医薬品に対して、バイオシミラーの品質特性がまったく同一であるということを意味するのではない。バイオシミラーは品質特性において、先行バイオ医薬品と比べて類似性が高く、かつ、品質特性に何らかの差異があったとしても、最終製品の安全性や有効性に有害な影響を及ぼさないと科学的に判断できることをもって同等性と判断している。
このガイドラインは、実は先述した、先行バイオ医薬品の製造工程の変更に伴ってその変更前後の同等性・同質性を担保するときに用いたICH Q5Eを用いている。つまり、バイオシミラーも先行バイオ医薬品の製造工程変更後の承認に用いたと同じガイドラインを用いているとのだ(図表2)。
図表2
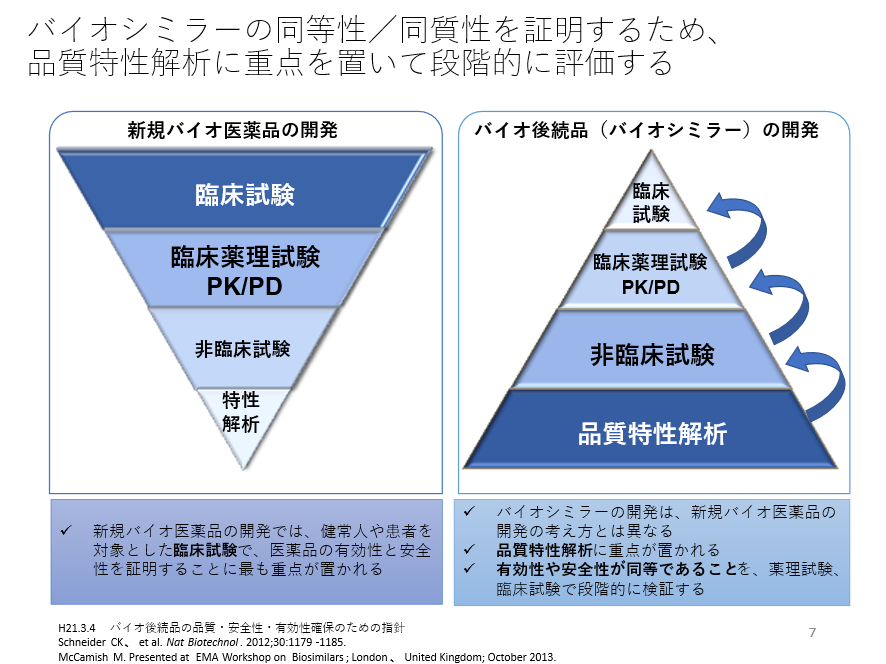
バイオシミラーの薬価は先行バイオ医薬品の薬価より安価だ。理由はバイオシミラーの開発経費が先行品の開発経費より安価だからだ。バイオシミラーの薬価は、先行バイオ医薬品のおよそ70%以下に抑えられている。なぜバイオシミラーの開発経費が安価なのかを見ていこう。それはバイオシミラーが先行バイオ医薬品より開発経費を抑えることが可能だからだ。実際に先行バイオ医薬品の場合、開発経費は1000~1800億円、開発期間も8~10年かかるのに対して、バイオシミラーはそれぞれ200~300億円で7~8年で済む。この理由は、先行バイオ医薬品の開発は、様々な候補物質をスクリーニングして臨床試験を通じて有効性、安全性を検証することで行われる。このため承認申請に必要な臨床試験の例数も800~1000人と多くの被検者が必要だ。
一方、バイオシミラーの開発は先行バイオ医薬品と開発の方向性が反対だ。バイオシミラーではすでに先行バイオ医薬品が存在するので、候補物質の品質特性解析に重点が置かれる。つまり先行品にできるだけ近い候補物質の品質特性解析を行い、その物質を用いて薬理試験、臨床試験へと開発が進む。つまり開発方向が逆向きである。こうした開発のことをリバースエンジニアリングと呼ぶ。このリバースエンジニアリングのため開発経費が少なくてすみ、またその臨床試験の例数も500例ほどで済ませることができる(図表3)。
図表3
厚労省 「バイオ医薬品とバイオシミラーを正しく理解していただくために」講習会資料より 2019年10月
このようにバイオシミラーの開発経費が先行バイオ医薬品より安価なため、バイオシミラーの薬価も先行バイオ医薬品の薬価の70%から40%台に抑えられている。実際に慢性関節リウマチの患者にエタネルセプトを使用する場合を見ると、先行バイオ医薬品の場合、年間薬剤費は108万円かかるところを、バイオシミラーでは66万円で済み、42万円も安価だ。同様に潰瘍性大腸炎の患者がインフリキシマブを使用する場合では、先行バイオ医薬品の場合年間48万円かかるところをバイオシミラーでは20万円ですみ、これも28万円ほど安価になる。
3 先行バイオ医薬品とバイオシミラーの違い
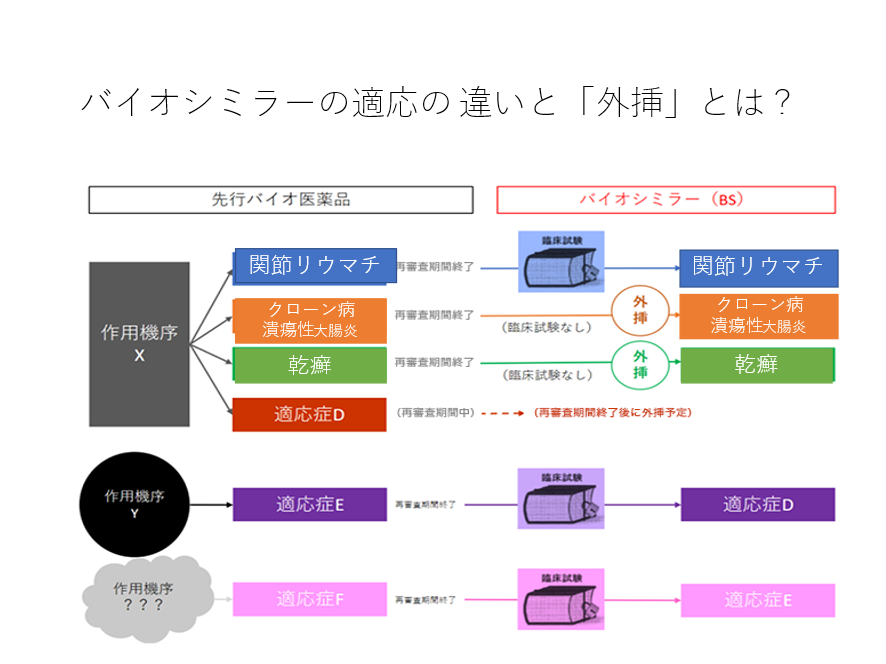
上記のようにバイオシミラーは先行バイオ医薬品より安価だ。しかしバイオシミラーは先行品バイオ医薬品との間には適応の違いの問題が存在する。つまりバイオシミラーは先行バイオ医薬品の持つ適応のすべて持っているわけではない。適応違いはなぜ起きるのだろう?
適応違いは実は特許の問題がからんでいる。医薬品の特許には物質特許、製法特許、適応特許の区別があり、適応特許は臨床試験を通じて適応が決められている。この適応特許が特許切れとならない限り、バイオシミラーは先行バイオ医薬品の適応のすべてを有することができない。適応特許は疾患によって特許取得時期が異なるので、先行バイオ医薬品の適応特許が切れていない場合は、バイオシミラーではその適応は認められない。こうしてバイオシミラーと先行バイオ医薬品の間に適応違いが生まれる。
しかしバイオシミラーには「外挿」という承認ルールが認められていて、先行バイオ医薬品の適応が治験なしでも認められることがある。外挿とは先行バイオ医薬品が複数の適応を有する場合、作用機序が同じ適応に関しては1つの適応について治験を行って、有効性、安全性が証明できれば、作用機序の同じ他の適応については治験なしでも承認を行うというルールだ。しかしバイオシミラーの場合でも、作用機序が異なる疾患については、その適応特許を取るためには、改めて治験を行い適応を承認を取得しなければならない。
たとえば、先行バイオ医薬品「レミケード」のバイオシミラーである「インフリキシマブ」は、関節リウマチについては治験を通じて先行品と有効性、安全性が同等であることを証明されている。これにより関節リウマチと作用機序が同じクローン病、潰瘍性大腸炎、乾癬(かんせん)については臨床試験を実施せずに、適応が承認されている(図表4)。しかしベーチェット病に関しては作用機序が異なるために適応は認められていない。
図表4
4 バイオ後続品の普及の現状と普及目標
次にバイオシミラーの普及の現状について見ていこう。2025年6月時点で、国内で上市しているバイオ後続品は19成分である。しかしその普及率をみると成分ごとにばらつきがある。バイオ後続品がバイオ医薬品に占める割合が80%を超すフィルグラスチム、エポエチンアルファ、ダルベポエチンアルファのような成分もあれば、20%程度しか普及していないインフリキシマブのような成分もある(図表5)
図表5
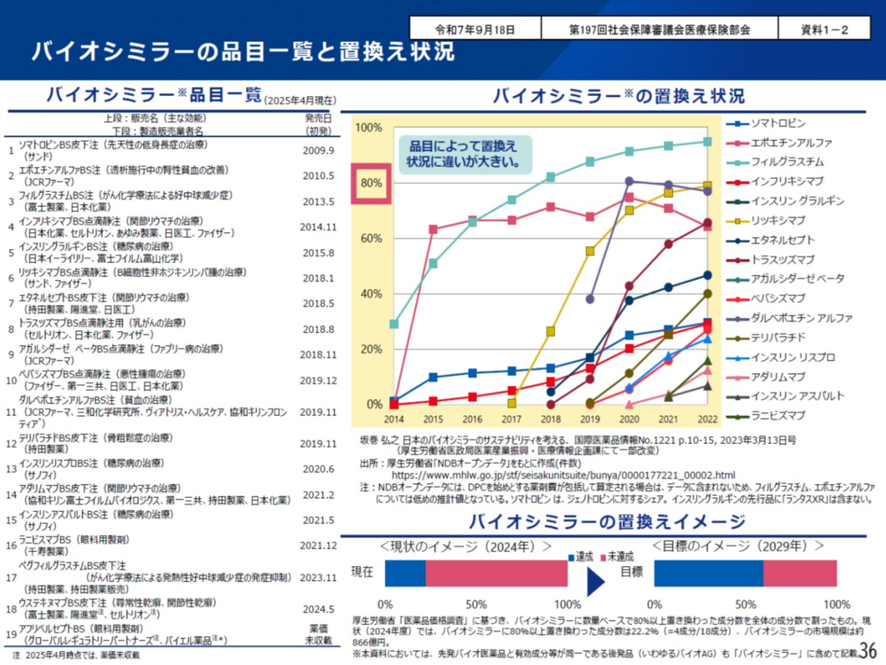
厚労省 中医協総会 2025年10月17日
さて国の医療費削減計画である第4期医療費適正化計画では、2029年度末までのバイオ後続品の普及目標を以下のように定めている。目標値はバイオシミラーが80%以上をしめる成分数が全体の成分数の60%以上としている。しかし現状では80%以上の市場シェアを占めるバイオシミラーは19成分中、先の3成分のエポエチンアルファ、フィルグラスチム、ダルベポエチンアルファしかない。このため現状では19成分のうちの3成分で、その割合は16%だ。これでは60%目標達成までには大分時間がかかる。というのも今後、先行バイオ医薬品が特許切れになると、バイオシミラーの成分数が増えていく。つまり分母が増えていくのだ。こうした中での60%目標だ。特許切れが多くなればなるほど、60%目標達成のハードルは高くなる。
5 2026年診療報酬改定とバイオ後続品
診療報酬でもバイオシミラーの普及促進を行う措置が行われている。普及促進を図る診療報酬上の仕組みとしてはバイオ後続品使用体制加算とバイオ後続品導入初期加算がある。バイオ後続品使用体制加算は、入院患者に対して、バイオ後続品の有効性、安全性について説明を行った上で、使用目標を達した場合に入院初日に点数を加算する。一方、バイオ後続品導入初期加算は、バイオ医薬品を使用する外来患者に対し初回投与から3か月間、月1回加点する。本加算の算定状況は年々増加している。
こうしたなか2026年の診療報酬改定では、さらなるバイオシミラー促進策が以下のように打ち出された。バイオシミラーの一般名を処方せんに記載した場合に処方せん料を上乗せするという「バイオシミラーの一般名処方」の新設だ。またすでにあるバイオシミラーを使用する保険医療機関に与える「バイオ後続品使用体制加算」の見直しを行う。また薬局でバイオシミラーを調剤した場合に、その割合に応じて薬局に加算を設ける「バイオ後続品調剤体制加算」も新設する。
以上のなかのバイオ医薬品の一般名処方の新設について以下に見ていこう。一般名処方ではバイオ医薬品を医師がその成分名である一般名を処方せんに記載し、「バイオ後続品でも可」とすれば、薬局側でバイオ後続品を選んで調剤することになる。実際にバイオ後続品には、一つの一般名に対して複数の企業が製品を上市している場合が多い。こうした場合に薬局側で複数ある製品のなかから1製品を選んで調剤するということになる。
たとえばインスリン、エリスロポエチン、フィルグラスチム(G‑CSF)、インフリキシマブ(抗TNF抗体)、リツキシマブなどは、1つの成分ごとの複数のバイオ後続品が存在する。たとえばインスリン製剤(インスリングラルギンなど)には先行品に対して複数のバイオシミラーがある。同様に腎性貧血の時に赤血球を増殖させるエポエチン(エリスロポエチン)にも複数のバイオ後続品がある。フィルグラスチム(G‑CSF)は化学療法の時に減少する白血球を増殖させるバイオ医薬品だ。このフィルグラスチムには複数のバイオシミラーがある。またリウマチやクローン病、潰瘍性大腸炎に使用するインフリキシマブ(抗TNFα抗体)にも先行品に対して複数のバイオシミラーがある。リンパ腫に用いられるリツキシマブ(抗CD20抗体)の先行品に対して複数のバイオシミラーがあるのが現状だ。
こうしたバイオ先行品に対して複数のバイオシミラーがある場合、医師がバイオシミラーの一般名処方を行い、「バイオ後続品でも可」とすれば、数あるバイオシミラーの中から特定のバイオ後続品を薬剤師が選んで調剤することになる。まさに低分子の後発品を一般名処方から薬剤師が選び調剤すると言う「変更調剤」の原則が、バイオ後続品にも適応されたということだ。
ただ低分子の後発品の一般名処方の場合は、わざわざ後発品でも可と医師がかかずとも、後発品を薬剤師は自動的に選ぶことができる。しかしバイオ後続品の場合、一般名処方に対しても「バイオ後続品でも可」と医師が指示する必要がある。これは低分子の後発品の場合、有効成分は先発品と後発品とは全く同一であるのに対して、バイオ先行品とバイオ後続品と有効成分は全く同一ではない、いわゆる「シミラー(類似品)」という扱いだからだ。
ただバイオ後続品の承認にあたっては先述したようにその物性の類似性、治験での有効性、安全性が確認されている。また後述するように先行品から後続品への切り替え時の有効性、安全性に関するスイッチング試験なども行われている。こうしたことよりバイオ後続品への切り替えについてエビデンスが積み重ねられた結果、今回のバイオ医薬品の一般名処方の解禁へとつながった。
6 バイオシミラーの切り替え試験とガイドライン
欧米ではバイオ先行品からバイオシミラーへの切り替え試験が行われている。有名な試験はノルウエーで行われたNOR-SWITCH試験だ。
NOR-SWITCH試験は、先行バイオ医薬品のインフリキシマブ(Remicade®)からバイオシミラー CT‑P13(Remsima®)へ「切り替えても問題ないか」を、 有効性・安全性・免疫原性の観点から検証した世界初の大規模・二重盲検・無作為化・非劣性試験だ。 ノルウェー政府が主導し、バイオシミラー切替政策の科学的根拠として極めて重要な試験となった。
NOR-SWITCH試験の概要は以下である。対象薬剤はインフリキシマブ(先行品)を CT‑P13(バイオシミラー)に切り替えた。対象患者は6つの炎症性疾患で6か月以上インフリキシマブで安定していた成人500名近くを2群に分けて、一方はバイオ先行品のまま継続、もう一方はバイオシミラーに切り替えた。
試験の主要評価項目は 52週以内の「病勢悪化(disease worsening)」の発生率で比較した。結果は病勢悪化の割合は先行品継続群で26%、CT‑P13(バイオシミラー)へ切替群で30%で、統計学的には非劣性(同等性)が示された。また安全性・免疫原性、有害事象、重篤な有害事象、抗薬物抗体の発生率は両群で同等だった。ただし疾患別の非劣性試験は疾患別の非劣性を検証する設計・検出力を持たなかった。つまり炎症性疾患全体集団としての非劣性は示されたが、クローン病、潰瘍性大腸炎、関節リウマチなどの疾患ごとの結論は慎重に扱う必要があるとされた。NOR-SWITCH試験は、それまでバイオシミラーへの切り替えは危険であるという従来の懸念を払しょくするのに役立った。
こうした切り替え試験もあり、欧米では、バイオシミラーの学会ガイドラインへの搭載が盛んだ。たとえば欧州リウマチ学会、欧州クローン病・潰瘍性大腸炎会議、欧州臨床腫瘍学会、米国臨床腫瘍学会ではバイオシミラーのガイドラインへの搭載や教育活動が盛んだ(図表10)。日本においても日本癌治療学会が顆粒球コロニー刺激因子(フィルグラスチム)の適正使用をガイドラインに記載している。
こうしたバイオシミラーの関連学会のガイドライン搭載が医師へのバイオシミラーの認識を高め、バイオシミラーの普及には欠かせない。
図表10

厚生労働省主催 講習会「バイオ医薬品とバイオシミラーを正しく理解していただくために」資料より
さて、バイオシミラーによる医療費節減効果はどれくらいだろう?バイオシミラーの薬価は先行バイオ医薬品の70%とされており、バイオ後続品全体の2024年度の医療費適正効果額は1,103億円に上る。バイオ医薬品の薬価が高いので、バイオ後続品による医療費節減効果は大きい。後発品による医療費節減効果額が1.6兆円と比べればまだ少ないほうだ。しかしこれから大型のバイオ先行品の特許切れの時代がやってくる。肺がんの抗がん剤であるオプジーボも2031年には特許が切れる。糖尿病薬のGLP-1受容体作動薬のリベルサスは2032年、糖尿病薬でヒット作品のマンジャロですら2036年に特許切れを迎える。
大型のバイオ先行品の特許切れがこれから続出する。これによりバイオ後続品の医療費削減効果がますます際立っていくだろう。
参考文献
- 厚生労働省医薬食品局審査管理課長 生物薬品(バイオテクノロジー応用医薬品/生物起源由来医薬品)の製造工程の変更にともなう同等性/同質性評価について 薬食審査発0426001号 平成17年4月26日
2)厚生労働省医薬食品局審査管理課長 バイオ後続品の品質・安全性・有効性確保のための指針 薬食審査発第0304007号 平成21年3月4日
3)内閣府「経済財政運営と改革の基本方針2017」平成29年6月9日